유교수님 광학 수업
빛은 입자일까 파동일까.
20251105-Optics.pdf
진동수, 시간에 대한 inverse
운동량, 공간에 대한 inverse
이 정도는 알자. 보라는 400nm 빨강은 700nm 수준이다 .
빛의 반사와 굴절.
굴절은 속도가 다른 매질을 지날 때 최단 시간으로 지나는 경로를 따라.
굴절률, 굴절율이 클 수록 느리다. 매질에서 광속을 진공에서 광속과 비교.
스넬의 법칙이 뭐더라!
식이 뭐더라, 이거다. 나 이제 유도할 수 있다. Snell’s Law
전반사
회절과 간섭
영의 이중슬릿 실험
하위겐스로 설명할 수 있죠?
편광
Polarization of Light
원형 편광 부분은 좀 봐두자.
원형 편광을 어떻게 만들 것인가?
두 진동의 위상차가 없으면 평면 위 편광이다.
위상차가 있으면 원형 아님 타원 편광이다.
x 성분과 y성분의 진폭이 같으면 원형 편광이다.
위상차에 따라 원형 편광과 타원 현광의 위상 회전 속도가 달라진다.
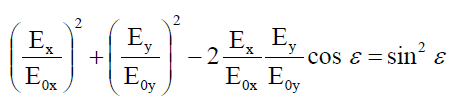
이 메인 방정식을 어떻게 유도할 수 있지? → 쉽다.
흡광
Jablonski diagram
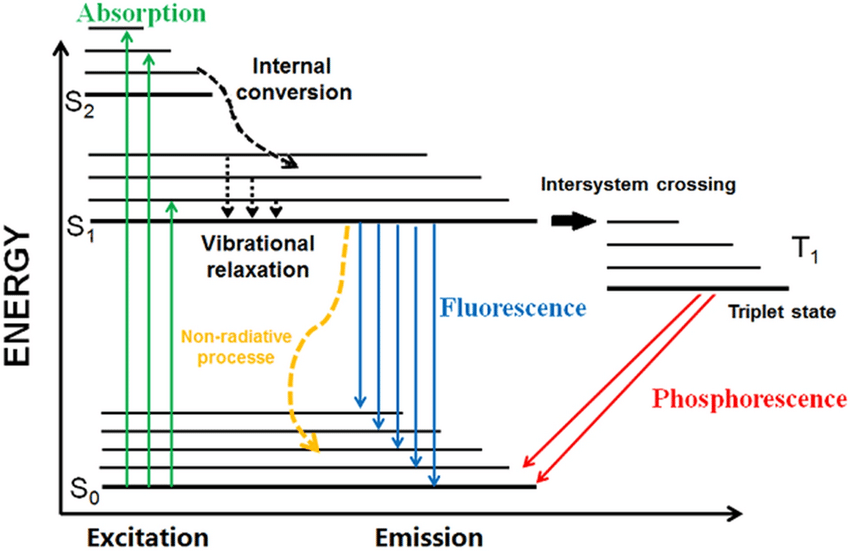
fluorescence
vibrational relaxation
Internal conversion(무복사 전이)
생체 분자의 빛 흡수와 색깔
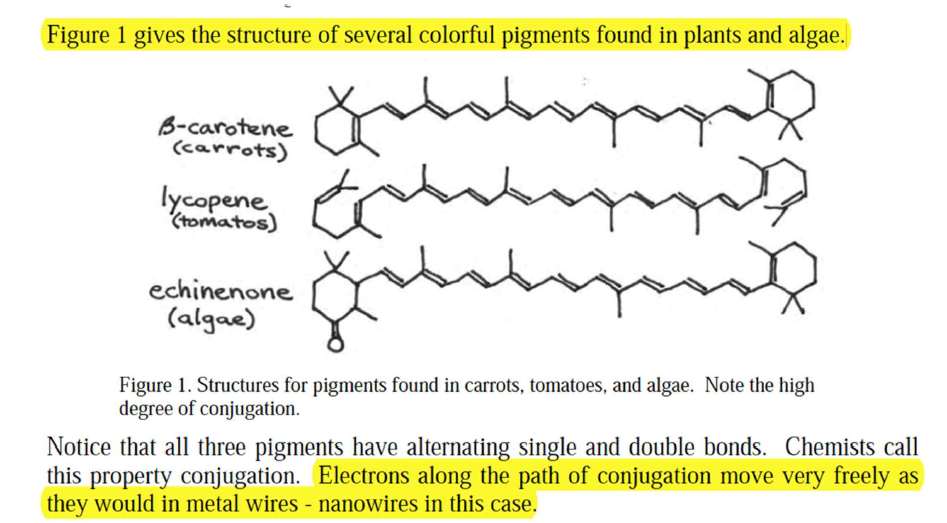
chemical conjugation에서 노니는 전자
단일 결합은 빙빙 회전할 수 있어요!
particle in a box
전자가 가질 수 있는 진동수는?
분자 크기 안에서 전자의 정상파

또는

이런 분자도 대충 박스라고 생각하고 슈방을 풀면 된다.
분자가 길면 긴 파장의 빛을 흡수할 수 있다.
클 수록 빨갛고, 작은 수록 보라.
벤젠링이 있는 아미노산도 가시광선 근처의 빛을 흡수할 수 있다.
Stoke’s shift
형광을 위한 들뜸 에너지는 형광으로 방출되는 에너지보다 적다. 흡수한 에너지의 일부는 vibrational relaxation으로 열이 되기 때문
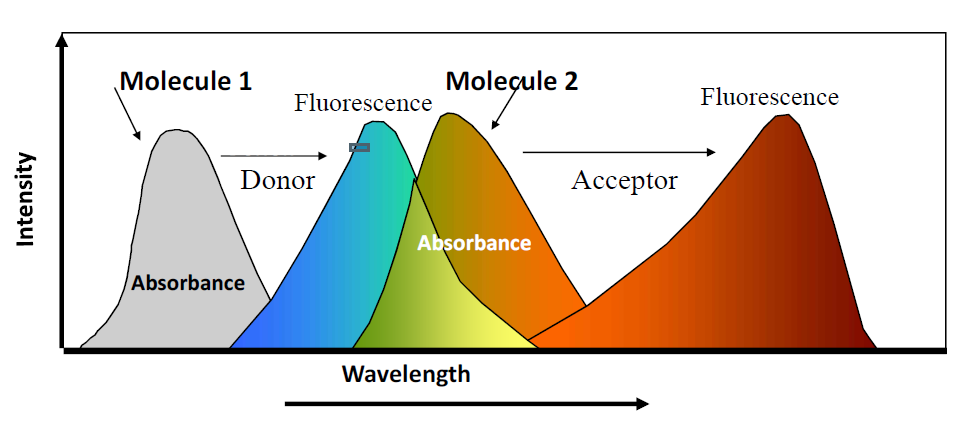
FRET
알죠?
그림을 보고 무슨 현상인지 맞춰라.
흡광과 형광의 차이.
형광의 가장 중요한 특징을 알아라. (Stoke’s shift)
이창훈 교수님 분광학 수업
Optics_실험1_분광학_2025.pdf]
생물리에 쓰이는 많은 장비에는 광학에서 나왔다.
18세기 쯤 빛에 대한 지식이 많이 쌓였다.
빛이 생명에 어떤 영향을 끼칠까?
비전, 광합성, 상 같은 용어라고 하던데요?
터비디티가 뭐죠? turbidity(탁도, 뿌연 정도)
빛이 산란되는 정도를 측정. 산란이 많이 되면 빛이 다시 측량되는 양이 줄어들겠죠? 세포가 많다.
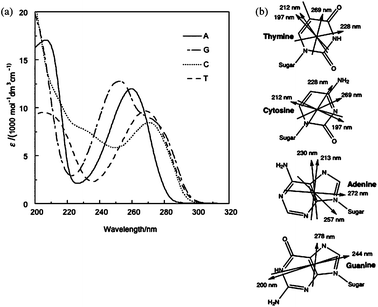
DNAbase에 따라 흡광하는 UV 파장대가 다르다.
두 베이스의 비율을 이용해, 염기 서열(GC ratio)를 알고 있다면 흡광 스펙트럼을 정확하게 계산할 수 있다.
펩타이드 본드 자체도 conjugate 구조를 가지기 때문에(conjugate 구조는 전자를 위한 박스) 빛을 흡수할 수 있다.
빛의 파장과 전자 구름의 크기는 어느 정도 비례
박스가 클 수록 큰 파장, 낮은 에너지.
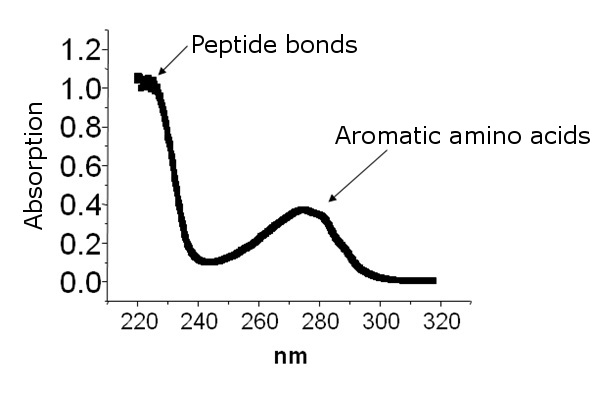
펩타이드 본드의 에너지가 아로마 링 보다는 에너지가 크다.
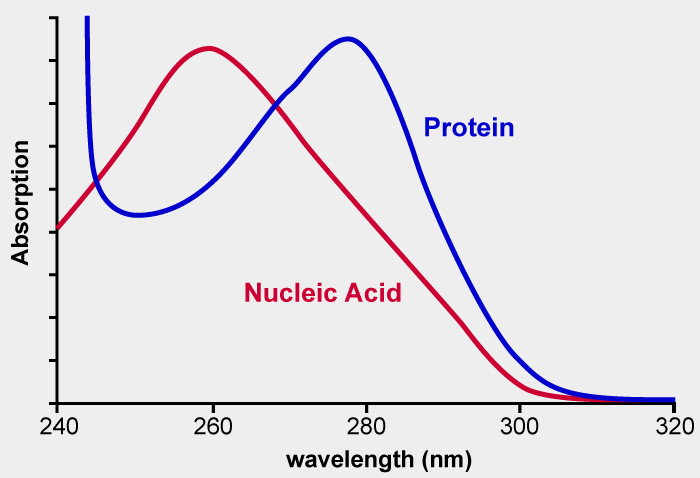
단백질은 280에서 피크, 핵산은 260에서 피크
이 비율을 구해서 정제가 잘 되었는지 확인할 수 있다.
흡광도
투과도(transmittance)는 output/input. 즉 쪼인 빛과 투과된 빛의 비율
10^-A 라고 지수로 나타낸다.
이때 A가 흡광도 absorbance라고 하자.
상용 로그다.
이렇게 지수로 나타내면 농도에 대한 함수로 나타내기 쉽다.
흡광도 = 소멸 계수 x 흡수체 몰농도 x 시료 경로 길이(cm)
소멸 계수와 시료 경로 길이를 알고 있다면, 흡광도를 측정해서 농도를 계산할 수 있다.
물론 extinction coefficient는 파장에 따라서 달라진다.
absorbance spectrum이라고 부른다.
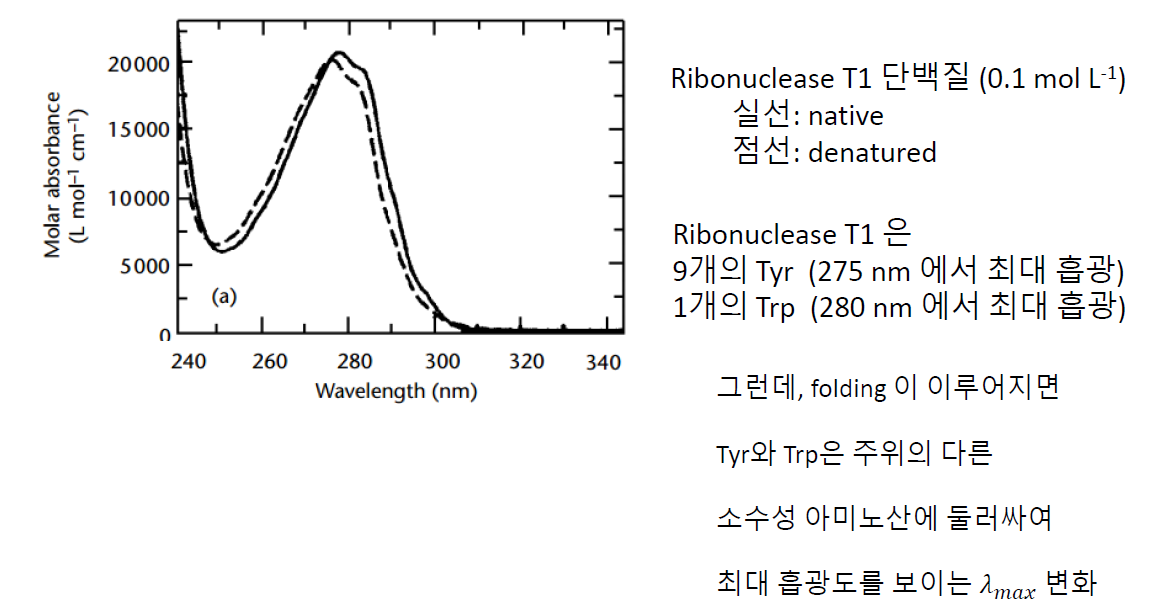
파장을 달리해 가지고 흡광도를 측정하면, 분자의 상태를 알 수 있겠네?
실제로 단백질이 변성되면 그래프가 이동한다. 교수님은 이걸 적색 편이라고 부르시더라.
분자의 상태가 차차 변화해도 특정 파장대에서는 흡광도가 똑같다.
아래 그래프에서는 pH가 단백질의 변성을 야기한다.
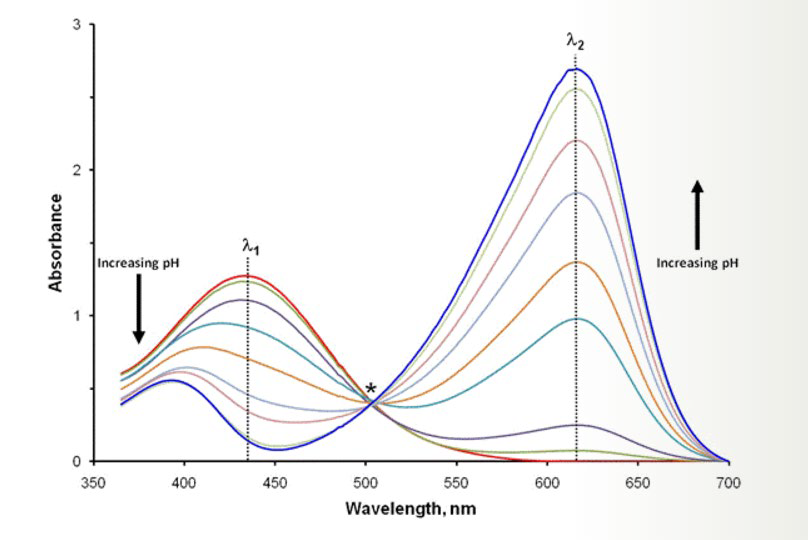
이걸 등흡광점(isosbestic point)이라고 부르자.
이 등흡광점을 기준점 삼아, 다른 파장대에서 흡광도가 달라지는 정도를 표준화할 수 있다.
그러면 두 가지 상태의 비율을 알 수 있다.
어떤 시료의 absorvance는 두 가지 상태의 고유한 extinction coefficient와 두 가지 상태의 비율에 의해 영향을 받을 터.
(아래 식에서는 빛의 진행 경로에 대해 표준화한 모양)
만약 특정 파장(
즉,
이렇게
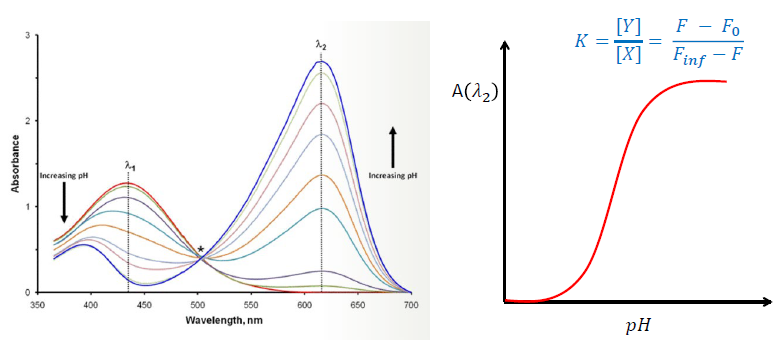
이렇게 구한 두 상태의 비율은 ph와 pK의 합으로 분석할 수 있다.
H는 high pH에서 단백질의 상태를 나타내고, L은 low pH에서 상태를 나타낸다.
absorbance A가 pH가 높아짐에 따라 높아진다면, 아래와 같이 수식을 세울 수 있겠다.
이 식을 거꾸로 알고 있었다. 하마터면 큰일 날 뻔 했다.
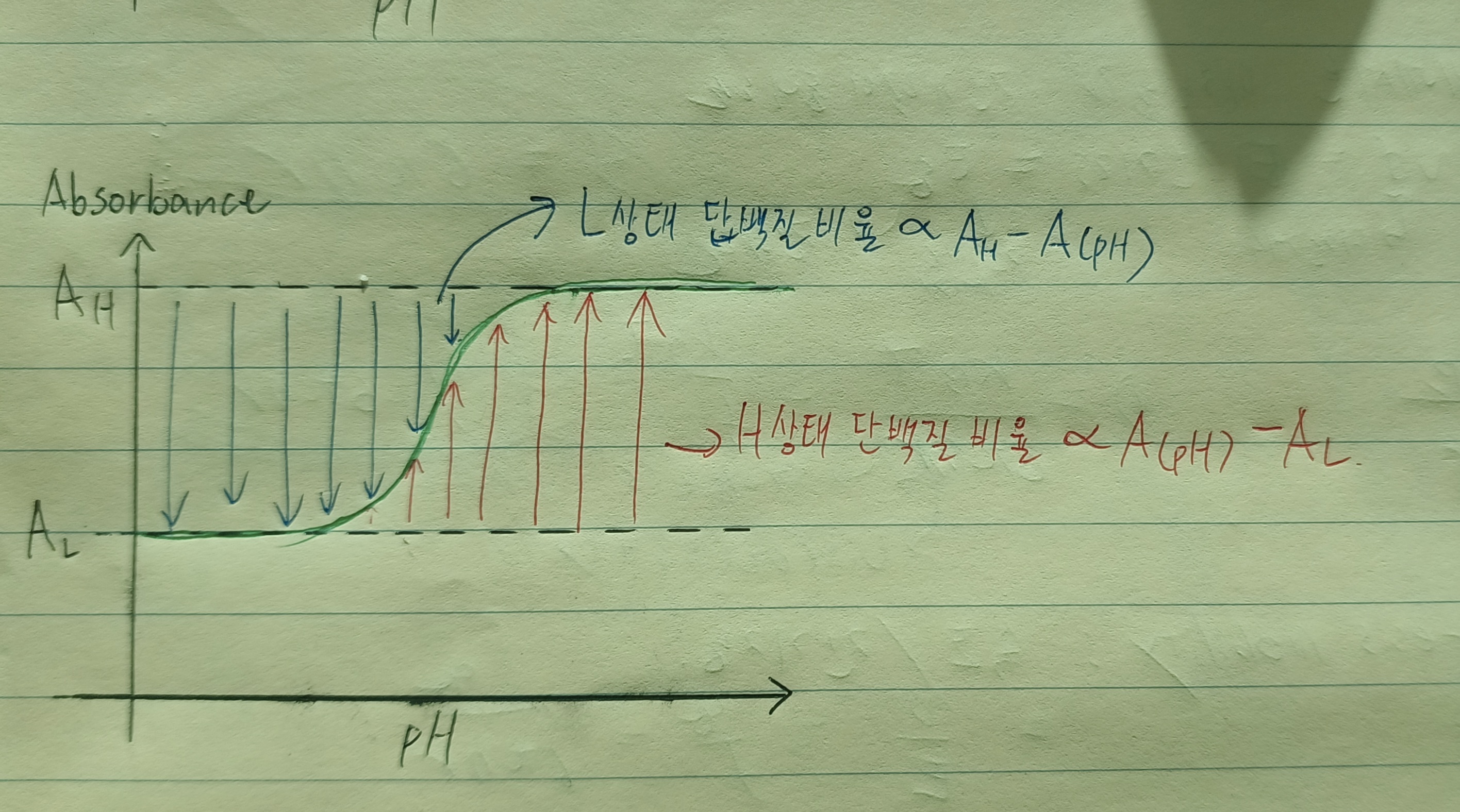
높은 pH에서 H+ 가 농도가 낮으니까, 단백질에서 H+ ion이 떨어져 나간다. 단백질은 다음 반응식을 따른다.
따라서 K의 정의는
앞서 흡광도로 구한
이렇게, 좌변을 pH에 대한 그래프로 그린다면 y 절편이 pK가 된다.
시험 문제 나온다!!!
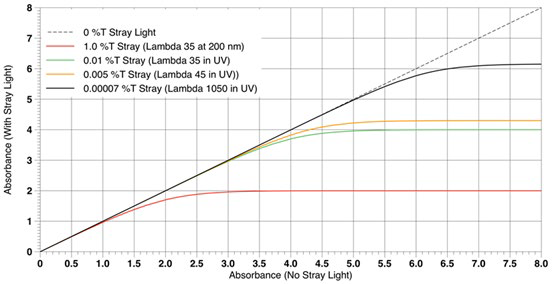
흡광도에는 한계가 있다. stray light에 의한 한계. stray light가 센서에 들어가서 정확한 absorbance보다 측정값이 낮게 나옴.
무슨 말인지 제대로 못 알아들었다.
흡광도가 커지면 커질 수록 선형성을 잃어버린다.
시료가 너무 고농도일 경우(흡광도가 너무 커질 경우) 농도를 낮추어 측정해야 한다.
이게 아직도 해결이 안 된 문제. 산란된 모든 빛을 모을 수 있도록 센서를 설치하기에는 무리.
UV-visible spectroscopy
빛의 형광
두 분자가 얼마나 가까이 있는지 알 수 있다.
FRET이랑 그 이름 어려운 거…
Fluorescence Protein Complementation
fluorescent spectroscopy
빛의 편광
circular dichroism
CD를 찍는 파장대에 따라 볼 수 있는 게 다르다.
긴 파장이라면 alpha helix의 떨림
아주 짧은 파장이라면 원자 단위로
200nm아래에서 데이터는 버퍼때문에 노이즈가 많이 낀다.
단백질의 alpha helix와 betasheet 의 CD 스팩트럼을 알고 있으니,
시료의 스펙트럼을 둘의 선형 합이라고 가정하면
alpha와 beta의 비율을 알 수 있다.
CD는 helix의 left와 right를 특히 잘 구분하겠지. 스팩트럼이 대칭형으로 나타난다.
아밀로이드에서 beta sheet가 쌓이면서 나름의 회전이 생긴다. 이로 인해 CD에 뭐가 보이지 않을까?
Fluorescence anisotropy
형광의 편광 각도까지 측정해보자.
그런데 보기 힘들어요… 작은 단백질은 보기 힘들어요
떼끄놀로지아~
빛의 회절과 산란
x선 회절
피크가 일어나는 각도를 통해 입자 사이 거리를 알 수 있다.
심지어 격자가 배열된 각도를 알 수도 있다.
SAXS가 뭐의 약자이지요?
small angle X-ray scattering
여기서 먼 거리일 수록 small angle. 각도와 시그널이 나오는 각도는 역수이다.
wide angle은 WAXS
어떤 분광계를 이용하면 뭘 볼 수 있을까?
이창훈 교수님 현미경 수업
지금은 바이러스, 100nm정도 볼 수 있는 단계.
MRI도 해상도를 현미경 수준으로 높일 수 있을지도?
near field microscopy의 대표 주자 - AFM (atomic force microscopy)
팁을 원자단위로 만든다면 원자 하나 단위의 세부 사항을 볼 수 있을 정도?
해상도의 정체 구간은 원자 사이 interaction
파장이 짧은 전자 빔, 높은 해상도.
전자 말고 쏠 쑤 있는 광원이 뭘까? x선 수준, 그 이상의 방사선을 쏘면? 분자가 무너지고..
그리고 굴절을 시킬 수 없다. 너무 파장이 짧으면 매질을 통과해도 위상 지연이 안 생겨.
상이 맺히는 방식과 굴절 방식에 따른 현미경의 종류
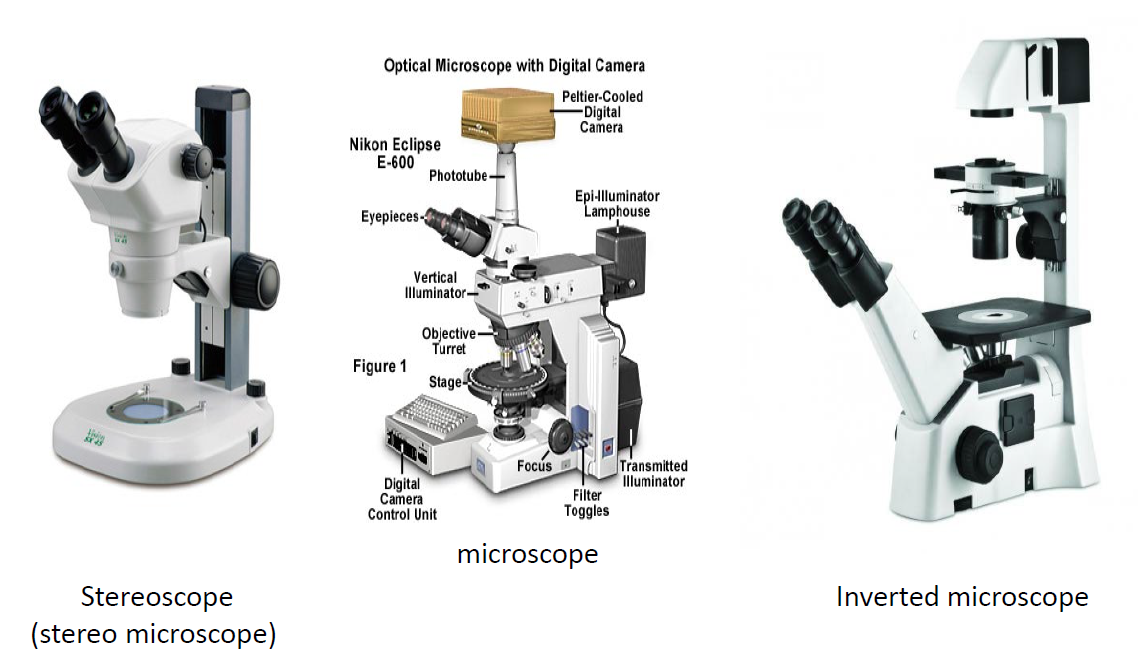
inverted microscope: 렌즈가 플레이트 아래에. 세포 보기 좋음. 초점이 맞는 단면을 볼 수 있음
microscope: 렌즈가 플레이트 위에. 전통적. 거울을 광원으로 쓸 수 있음. 초점이 맞는 단면을 볼 수 있음(2)
stereoscope(해부현미경): 해상도가 낮고 입체 구조를 볼 수 있음. 세포보다 큰 걸 볼 수 있음.
phase contrast microscope: 빛의 위상을 이용. 특정 위상을 걸러내는 필터가 있다. 이러면 상의 대비가 높아진다. 위상차에 따라 여러 이미지를 얻는 것도 가능.
differential interference contrast(DIC): 위상을 아주 세부적으로 골라낸다. 광원에서부터 위상을 골라서 쏜다. 프리즘이 앞과 뒤 두 개 있다. 갈라진 두 빛을 합쳐주면서 간섭현상이 일어난다. 세포 막의 주변부가 뚜렷하게 보인다. vesicle같은 걸 볼 수도 있다. 이렇게 좋은 대신 비싸다.
형광으로 라벨링해서 보기
다른 빛의 간섭을 받지 않기 때문에 해상도가 높다? 무슨 말이지? 더 자세히 알아봐야지?
형광 세기의 비표: 양자 수율 = 방출된 광자 수 / 흡수된 광자 수
EGFP는 원래 GFP에 돌연변이를 일으켜서 단백질이 더 안정된 상태, 형광을 더 잘 뱉는다.
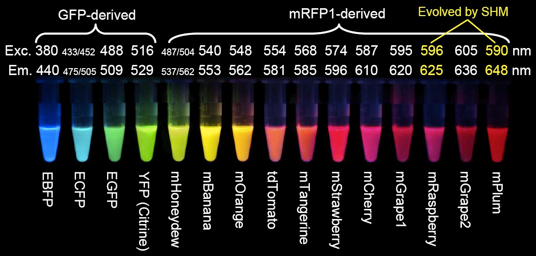
형광 이름이 왜이리 맛있게 보여요.
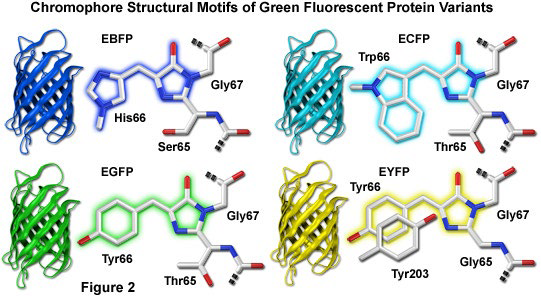
아미노산을 다르게 하면 에너지가 약간씩 바뀌고 색이 달라져요.
형광단백질은 beta barrel안에 진짜 형광을 내는 구조가 갇혀있는 모양.
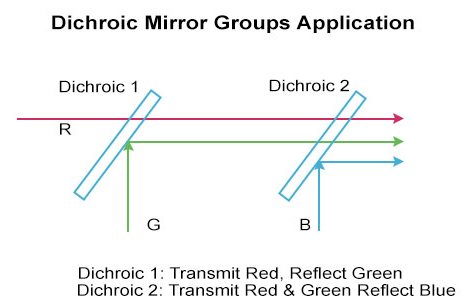
형광 현미경에 있는 dichromic mirror. 장파장은 통과시키고 단파장은 반사하는 등.
가시광선 영역에서 단파장은 굴절률이 크니까 전반사가 더 잘 일어나는 원리인가?
염료에 맞는 파장만 골라서 쏴준다.
광원이 필터를 통과하면서 생기는 문제.
다양한 파장이 섞여있어 색수차때문에 초점이 안 맞는다.
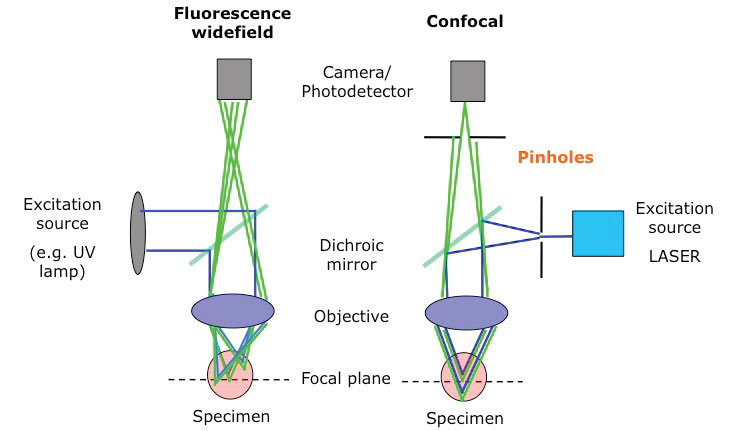
confocal: 단 파장의 레이저만 쏘자. 초점이 아주 잘 맞는다. xyz 모든 방향에서 초점을 맞출 수 있다. 사진 여러번 찍으면 3차원 구조 볼 수 있다. 단점, 고에너지라서 시료가 탄다.
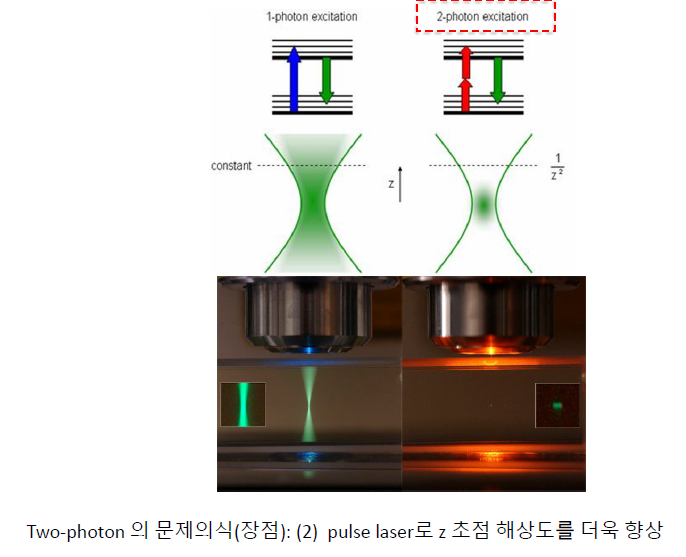
형광이 탈색이 된다. 그렇다고 긴 파장을 쓰면? 형광이 안 보이지 않나? 그럼 two step으로 excitation시켜보자. 이게 왜 양자적으로 되지?
two step이라서 빛을 두 번 쏘여야 해. 이러면 한정된 공간에서만 excitation이 일어나도록 강요할 수 있음. z 축 해상도가 높아짐.
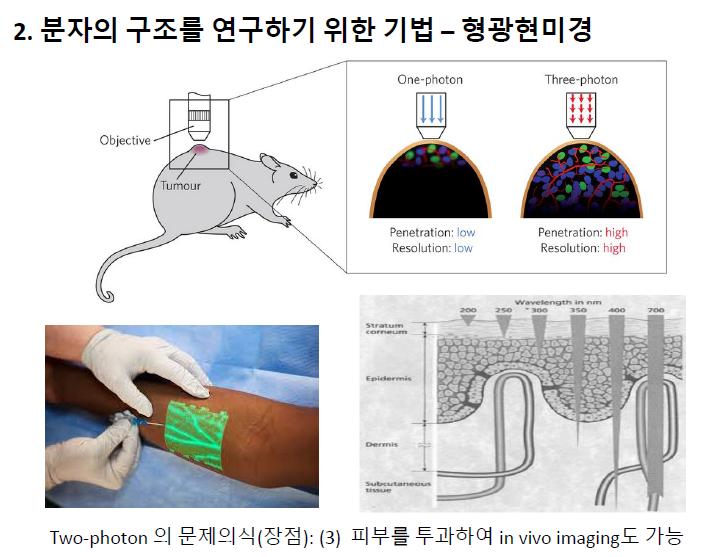
가시광선 영역에서 파장이 길면 투과가 잘 된다. two-photon은 피부를 잘 투과한다.
긴 파장을 쏘니까 빛이 안쪽까지 잘 들어간다. 혈관까지 잘 보여요.
정리, two-photone method의 장점
- 형광을 발하기 위해 원래 필요한 빛보다 장 파장을 주어 시료가 타지 않아
- 2번 빛 주는 타이밍을 조정하여 z축 해상도를 높일 수 있어
- 피부 투과도가 높다.
광학 현미경의 한계
아벨의 공식. Abbe theory
빛 굴절의 한계
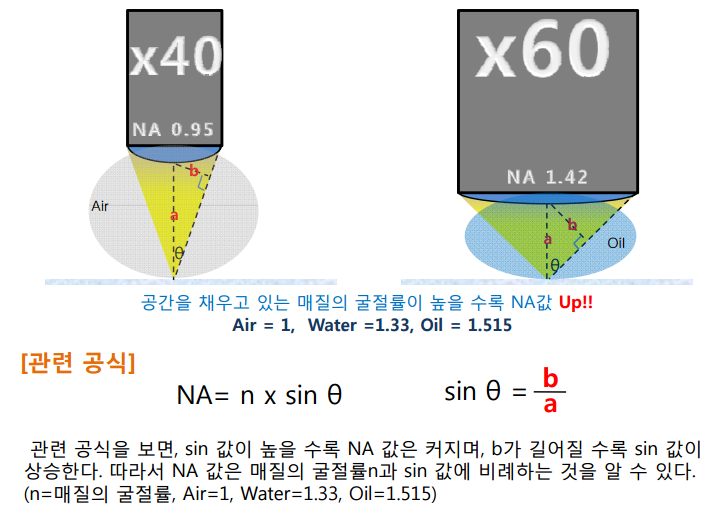

numerical aperture(개구수) 렌즈가 얼마나 많은 빛을 한 곳으로 모을 수 있나?(얼마나 잘 굴절시키나?)
렌즈 배율이 높으면 초점이 맞는 z축의 폭이 좁아진다. (관찰 두께가 얇아진다.)
이 한계를 깨자, super resolution
STED(stimulated emission depletion)
와 아는 거다
SIM(structured illumination microscopy)
슬릿을 이용.
localization microscopy, PALM(Photoactivated localization microscopy), STORM( Stochastic Optical Reconstruction Microscopy)
시간차를 주어서 여러 위치에 형광을 켠다.
holographic tomography(잘 모름, 찾아봐야 함. )
더 입체적으로 보자. 표지가 필요 없다. 대신 컴퓨터를 혹사시킨다.
토모그래피. 표지 없이도 vesicle정도는 볼 수 있다.
굴철 차이를 보기 때문에, 빛이 통과한 영역의 굴절률을 알 수 있고, 이게 물인지 지질인지 단백질인지 알 수 있다. 혁명적이군. 그런데 해상도가 confocal을 넘지 못했다.
전자 현미경
전자의 물질파는 드브로이 물질파 공식
전압으로 전자를 가속.

전자현미경으로는 표지로 금속을 쓴다. 생체 물질은 전자가 밀집된 원자가 많지 않아서 화질이 좋지 않다.
전자 현미경도 입체적으로 찍자.
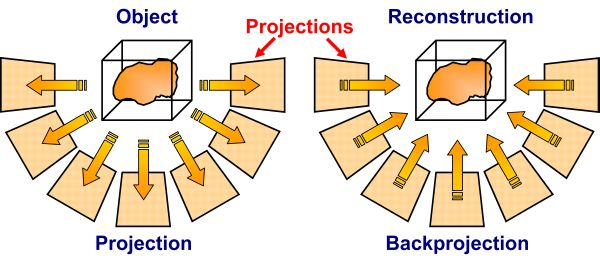
electro tomography
하지만 360도 돌릴 순 없다. 전자가 다 통과하는 각도면 센서가 타버려.

cryo EM 여러 각도로 얼어버린 시료를 볼 수 있어서 여러 각도의 모습을 모을 수 있다.
유교수님 NMR
20251117-NMR.pdf
nucleus magnetic resonance
핵 자기 공명
NMR 스펙트럼에서 관찰할 수 있는 네 가지 주요 특성과 그로부터 얻을 수 있는 정보는 다음과 같다.
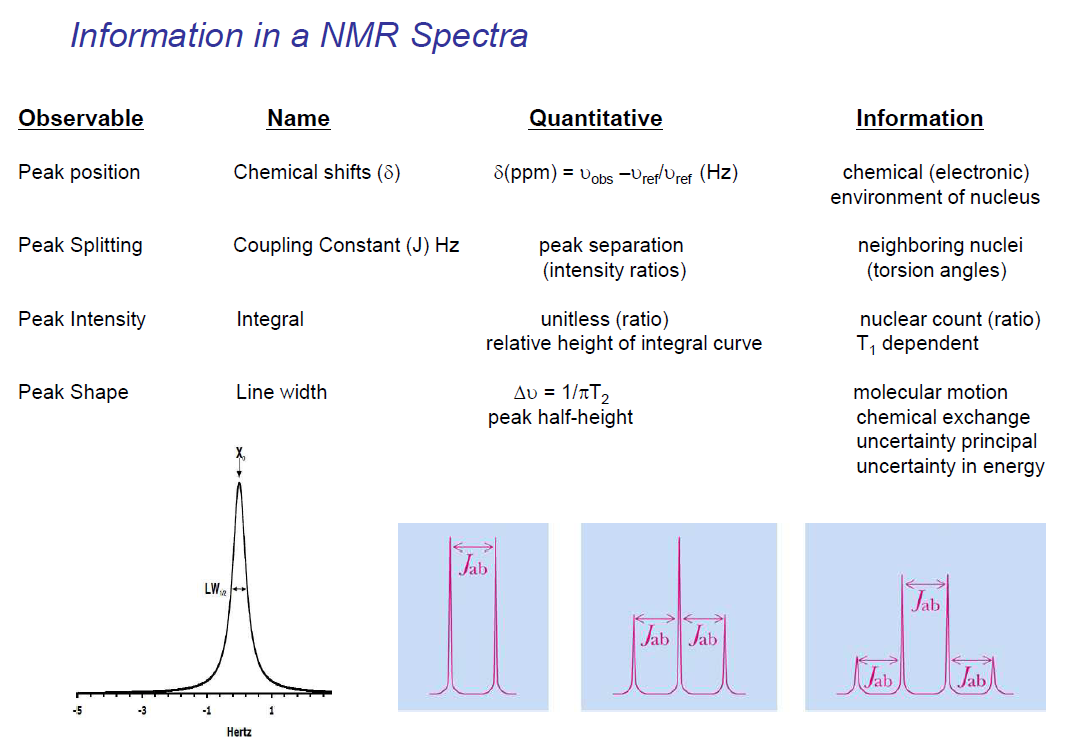
1. Peak Position (피크 위치)
Observable: Chemical shifts (δ)
정량적 표현:
제공 정보:
- 핵의 화학적(전자적) 환경
- 주변 전자 밀도와 차폐 효과에 따라 공명 주파수가 달라짐
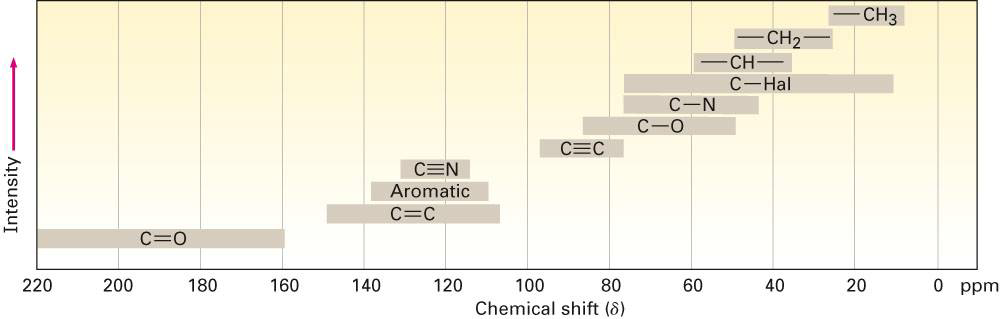
- 이렇게 같은 탄소여도 어느 작용기의 탄소인지에 따라 chemical shift가 달라진다.
- 작용기와 화학 구조 파악에 핵심적
- 근처에 electro negative한 원자가 있으면 chemical shift가 더 심해진다. 전자가 벗겨져서 차폐 영향이 사라지므로 외부 자기장에 더 취약해져서 업스핀과 다운 스핀의 에너지 차이가 더 커지니까?
- 탄소의 경우 결합 수에 따라 혼성 오비탈 구조가 달라진다. 이것또한 영향.
해석:
- 전자가 많이 차폐할수록 upfield (낮은 ppm, 원래보다 chemical shift를 덜 한다)
- 전자 끄는 그룹 근처일수록 downfield (높은 ppm)
- 예: 방향족 영역 (7-8 ppm), 알킬 영역 (0-3 ppm)
2. Peak Splitting (피크 분열)
Observable: Coupling Constant (J) Hz
정량적 표현:
- 피크 간 분리 거리 (intensity ratios)
- J 값은 Hz 단위로 측정, 자기장 세기와 무관하게 차이가 나기 때문
제공 정보: 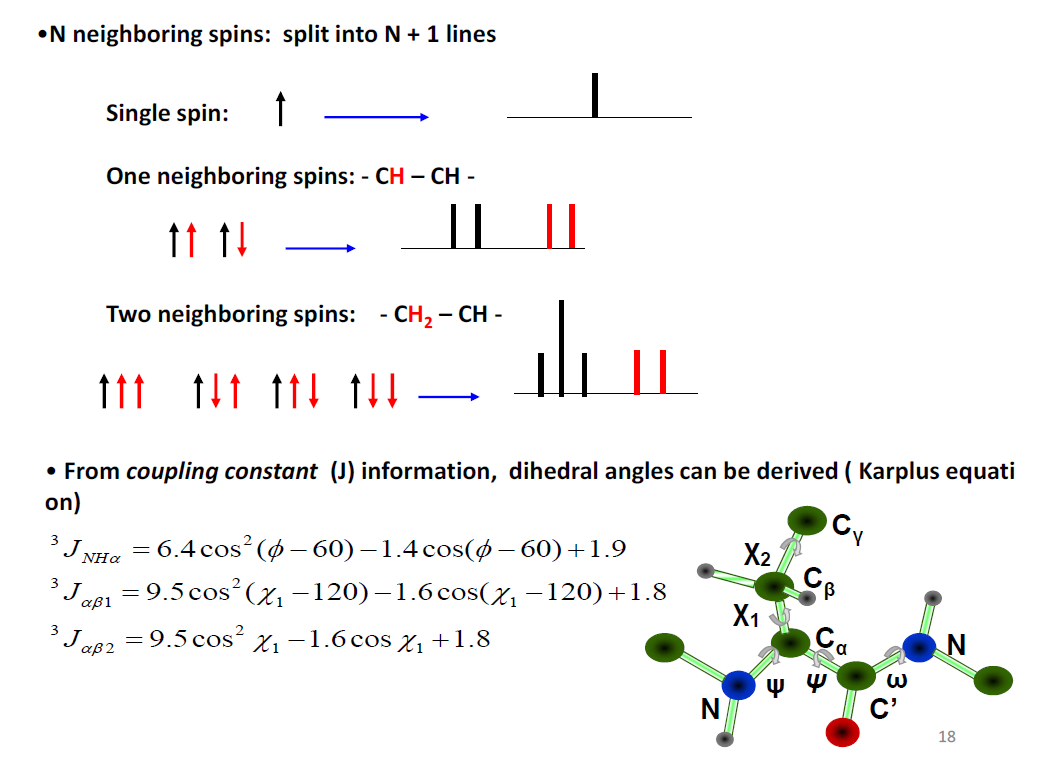
- 이웃한 핵들과의 스핀-스핀 결합
- 비틀림 각도(dihedral angles) 정보, 비틀림 각 달라지면 분령된 피크 사이 간격이 달라짐
- Karplus 관계식으로 이면각 계산 가능
- 해석:
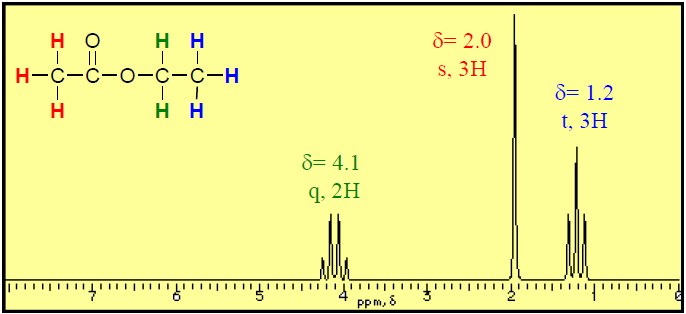
- n+1 규칙: n개의 등가 이웃 수소(같은 탄소가 아닌이웃 탄소에 붙은 등가 수소 개수) → n+1개로 분열
- 왜? n개 이웃이 만들 수 있는 총 스핀 합의 가짓수가 n+1이기 때문
- Doublet (d): 이웃 수소 1개
- Triplet (t): 이웃 수소 2개
- Quartet (q): 이웃 수소 3개
- Multiplet (m): 복잡한 분열 패턴
3. Peak Intensity (피크 세기)
Observable: Integral
정량적 표현:
- Unitless (ratio)
- 적분 곡선의 상대적 높이
- T₁ dependent (종방향 이완 시간에 의존)
제공 정보: - 핵의 개수 비율
- 정량 분석 가능
주의사항:
- ¹H-NMR에서 가장 정확
- ¹³C-NMR은 T₁ 차이와 NOE 효과로 정량 부정확
- 충분한 relaxation delay 필요
- 적분비는 정수비로 근사
활용:
- 화합물의 순도 확인
- 혼합물의 조성 비율 결정
- 구조 검증
4. Peak Shape (피크 모양)
Observable: Line width (선폭)
정량적 표현:
제공 정보:
- 분자 운동 (molecular motion)
- 화학적 교환 (chemical exchange)
- 불확정성 원리 (uncertainty principle)
- 에너지 불확정성
해석:
좁은 피크 (Sharp peak):
- 긴 T₂ (횡이완 시간)
- 빠른 분자 운동 (작은 분자, 낮은 점도)
- 균일한 환경
- 교환 과정 느림
넓은 피크 (Broad peak):
- 짧은 T₂
- 느린 분자 운동 (큰 분자, 높은 점도)
- 불균일한 환경
- 빠른 화학적 교환
- 중간 속도의 동역학 과정
응용:
- 단백질 크기 추정
- 결합 상태 vs 자유 상태 구별
- 동역학 연구 (microsecond-millisecond 시간대) - 온도 의존성 연구
핵자기란 뭔가? Zeeman effect
원자핵, 입자가 가진 고유한 스핀이 자기장을 만든다.
보통은 +1/2 아니면 -1/2
스핀이 아무렇게나 정렬된 것이 기본 상태.
외부 자기장이 주어지면 spin이 alighn 된다.
자기장이 가해질 수록 두 스핀의 에너지 차이가 나타난다.
자기장에 의한 energy level split, 이것이 바로 Zeeman effect(제이만 효과)인 것이다.
자기장을 쭉 걸어주면,
쐰 빛의 에너지와 같은 에너지 레벨 차이가 생기는 그 순간 빛이 흡수된다.
chemical shift
에너지 차이로 흡수하는 빛의 진동수가 얼마나 달라지나?
기준으로 삼을 기준 분자의 진동수와 비교하여, 진동수 차이를 확인한다.
이러면 어떤 자기장에서 실험하든, 상대적인 진동수 차이는 일정하다.
수소가 어느 분자에 있느냐에 따라서 chemical shift가 달라진다.
단백질 보는 사람은 펩타이드 결합, 아로마링,
탄소의 경우에도 탄소와 연결된 본딩에 따라 shift가 다르다.
NMR data의 x축 데이터는 뭘가?

ppm단위(
j-coupling
H가 여러개 인근해 있으면, 서로의 스핀에 의해 에너지 상태가 달라진다. 2개의 수소가 있으면 1:2:1 의 에너지 레벨이(??) 나뉘어 지는 거다.
수소가 3개가 있으면 1:3:3:1 이되는 거다.
그래서 peak가 분리된 형태를 보면 원자가 어떻게 붙어있는지 확인할 수 있다.
단백질 입장에서는 dihegral angle과 밀접한 관련이 있다.
nuclear Overhauser effect(NOE)
peak이 saturation되면서 없어지는 현상.
이 원자하고 저 원자하고 가까이 있다?
무슨말하는지 모르겠다!!!
핵 사이 거리 측정 도구라는 것만 일단 알아두자
왜 수소하고 질소를 한 쌍으로 보는 걸까요?
Hetero nuclear single quantum coherence는 뭐죠? 고급 개념이라고 하네요.
펄스로 자화가 xy 평면으로 넘어가고 나서 다시 z축으로 회복되는 시간
→ 이게 클 수록 peak intensity가 커진다.
xy 평면에서 신호 감쇠 시간
→ 이게 클 수록 peak width가 좁아진다.
NMR은 무엇에 쓰는가?
단백질 구조를 어떻게 보는가?
간접적인 단서
chemical shift로 2차 구조를 추정할 수 있다. → 왜 2차구조인진 모르겠으나 일단 납득하자.
J coupling으로 dihedral angle을 볼 수 있다.
NOE로 원자 사이 거리를 볼 수 있다.
residual dipolar coupling으로 본딩 각도를 볼 수 있다.
relaxation rates(T_1, T_2)로 macromolecular dynamics를 볼 수 있다.
이런 NMR의 구조가 나올 법한 단백질의 형태를 계속 시뮬레이션한다.
NOE가 많을 수록 단백질의 형태를 유추하기 쉬우며, 조금만 시뮬레이션해도 답을 찾을 수 있다. 솔루션속에서 단백질이 있을 수 있는 형태의 집합을 구할 수 있다.
NMR은 단백질의 크기가 작을 때 강하다! NH로 라벨링하는 경우는 그렇다.
Xray와 상호보완적으로 사용하면 좋다.
protein dynamics
단백질의 상태가 2가지 있으면, 이것이 전환되는 속도를 NMR로 측정할 수 있다. peak가 몇 개 뜨는지를 확인하여서. 느리게 바뀌면 각 상태에 대한 peak이 따로 잘 보이고, 빠르게 바뀌면 중간 단계의 peak이 보인다.
H-D exchange도 볼 수 있다. D는 스핀이 없어서 피크가 안 보이므로.
대부분 H 신호가 exponential하게 decay한다.
빠르게 decay할 수록 용액에 노출된 취약한 부위의 원자이다.
접힌 상태와 풀린 상태의 공존.
리간드와 온도를 바꿔가면서…
NMR 그래프의 의미
NMR로 protein 다이나믹스를 본다는 게 무슨 의미인가?
와 이게 무슨 말인지 제일 몰랐는데 이걸 외우라고?
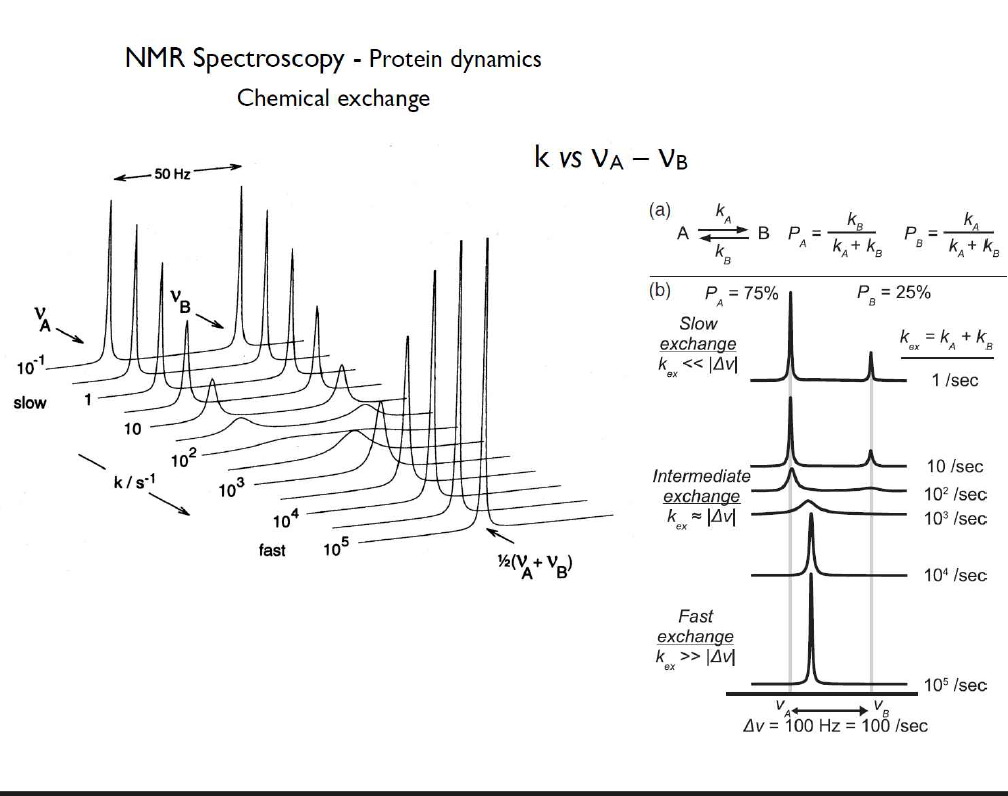
녹색에서 빨강이 되면 에너지가 낮아진다?
chemical shift의 크기가 뭔지 감이 안 잡히는데요.
Protein folding
20251119-Folding.pdf
풀린 상태에서 잘 접히는 그 과정을 실험으로 볼 수 있다면 참 좋을 텐데!
그럴 수 없어.
레비탈의 역설 levinthal’s paradox
포텐셜의 지리 Anfinsen’s dogma
왜 우리는 protein folding dynamics에 주목해야 하는가?
- 접히는 과정으로 넘어갈 수 있는 특정한 풀림 구조는 무엇인가?
- 어떻게 잘못 접혀서 병을 일으키는가? 오접힘의 과정을 스냅샷찍으면 좋겠다.
protein structure 연구의 세 가지 부분
- Native structure가 어떻게 생겼나? (접힌 구조가 어떻게 생겼나?)
- Denatured state가 어떻게 생겼나?
- protein folding kinetics(접힐 때 어떤 역학으로 접히나?)
Native state
보통은 여기에 많이 집중.
폴딩된 이후의 결과가 이것.
사용하는 장비 딱 정리
- X-ray crystallography, XRD (결정을 잘 만들어야 하는데 이게 어렵다)
- NMR(픽 하나 분석하는 게 시간이 많이 걸림, 작은 것만 볼 수 있다. ),
- cryo-EM (준비에 시간이 걸리지 찍는 건 빠르다. 큰 분자만 볼 수 있다. )
이런 걸 쓴다.
시뮬레이션으로는
- molecular dynamics simulation
- Monte Carlo simulation
Protein data base들
denatured state
이게 왜 중요하나?
랜덤하게 풀려있는 구조.
구조가 없는 게 구조인 단백질, insrasicaly disordered
사용하는 장비 딱 정리
- SAXS
- FRET
- CD (circular dichroism spectroscopy)
- NMR
SAXS로 단백질 크기를 측정하면 좀 크게 나오고,
(SAXS가 뭐였더라?)
FRET으로 측정하면 더 작게 나온다. FRET은 단백질의 크기를 재는 게 아니라 다이의 거리를 측정하니까…
2005년, 비정형 단백질이라는 개념이 생기다.
공학적 응용을 할 수 있어 중요한 단백질.
빛을 딱 쪼이면 폴딩되는 신기한,
바인딩 짝이 있으면 폴딩되는 신기한 단백질 .
풀리고 접히는건 대충 SAXS로 볼 수 있다.
CD로도 대충 볼 수 있다.
NMR로 볼 수 있다.
풀리면 주변 아미소산에 있는 chemical shift가 사라지기 때문.
IDR(insrinsically disordered proteins)의 역할이 뭔가
permanent binding
transient binding
no binding
대부분은 도메인 중 일부가, 혹은 터미널이 IDR일 수 있다.
타우, 알파시누클레인
LLPS 때문에 유명해진 그자체로 형태가 없는 단백질들.
Folding kinetics
단백질이 접혀가는 중간 과정.
- diffusion-collision model: 2차 구조 먼저, 3차 구조는 나중에
- hydrophobic collapse model: 물을 싫어하는 성질에 의해 3차구조의 대략적인 구조를 잡고 나서, 2차 구조가 생긴다.
누가 승자냐? 단백질마다 다르다.
딱 둘 중 하나로 나뉘는 것도 아니었다.
뭐? 허브 모델? Foldon funnel, zipping model?
sequentual stabilization model
funnel 하고 sequential stabilization model 하고 싸운대요.
- funnel model
- sequential stabilization model
sequential stabilization model은 현재 과정이 다음 접힘 과정을 더 안정적으로 만든다고
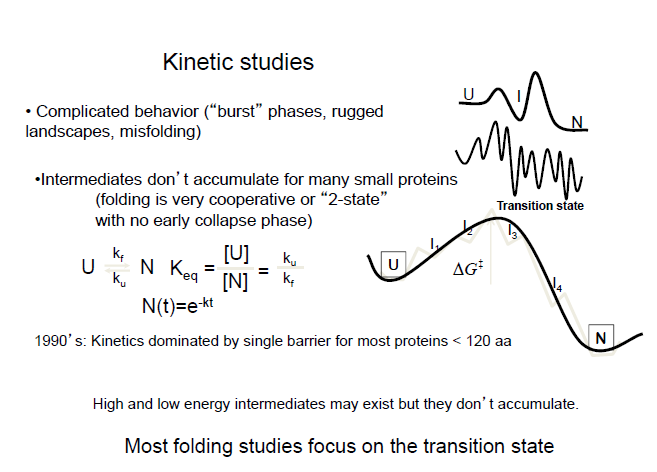
대충 2 state라고 두고 한번 풀어보자.
그래서 에너지 베리어
폴딩된 단백질과 Urea같은 단백질 풀리게 하는 버퍼를 점점 섞는다.
decay 시간을 보아, 빠르면 에너지 장벽이 낮은 거고, 느리면 에너지 장벽이 높은 거다.
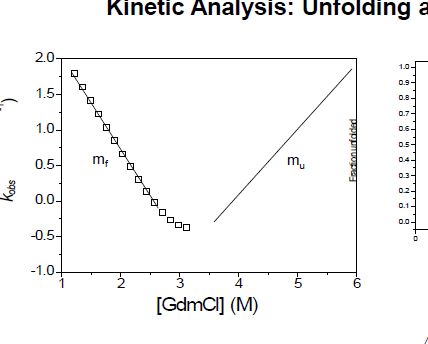
이렇게 V자로 나타낼 수 있다면, two state model을 쓸 수 있따.
transition state에 관여하는 아미노산을 어떻게 규정할까? mutation이 일어날 때 에너지 구조의 변화를 확인한다.
mutational pi-value analysis, Transition State Ensemble
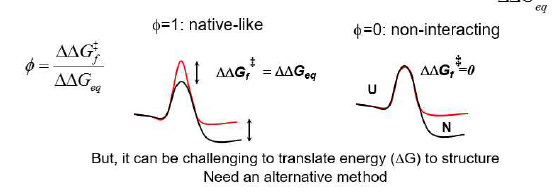
베리어 높이까지 올라가면 folding에 관여하는 거고,
베리어 높이가 안 변하면 folding에 관여하지 않는 것이다.
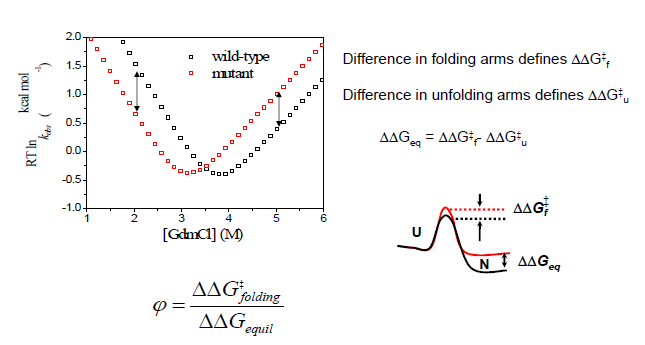
그런데 pi 값이 0.2 0.5 이런 애매한 값이면 어떻게 할 것인가?
==mutational psi-value analysis, Transition State Ensemble==
관심있는 두 아미노산을 His로 바꾸고, 아연을 넣으면? 값이 on and off로 잘 나온다.
이 값을 psi라고 부르자.
왜 이리 안 풀리지?
folding cooperativity
강의 뜻을 못 알아들았어.
단백질이 2차 3차가 확 풀렸다가 확 접힌다.
중간에 접혀가는 상태를 측정하기 상당히 쉽지 않다.
HDX(hydrogen exchange experiment)를 쓰면 그나마 준안정 상태를 측정하기 쉽다.
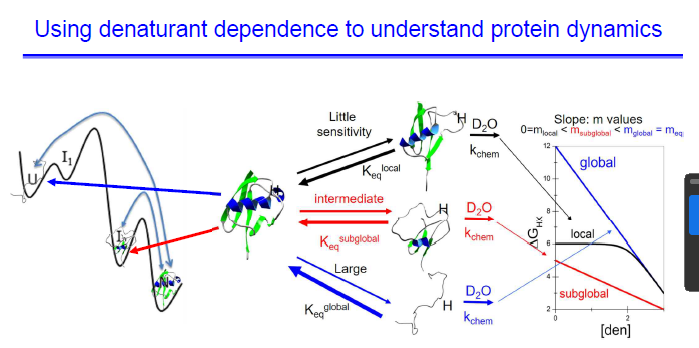
이게 무슨 내용이지
실험에서 본 단백질 역학이랑, 엄청 dense한 세포 내부에서 단백질 접힘이랑 같은 걸 어떻게 보장하죠?
다른 걸론 optical tweezer이나 AFM을 쓴다
시험 문제: 각 state를 연구할 수 있는 실험 방법하고 장치를 알아야 하지요
X선 결정학
2025_X선결정학_LMS.pdf
광물의 결정학과 우사하다.
그러나 단백질에서는 뭔가 좀 다르지.
격자에서는 원자들의 농도가 높을 수밖에 없다.
단백질이 일정하게 있으면 회절 구조가 나타날 수밖에 없다.
화절 무늬로 결정을 역추적할 수 있다.
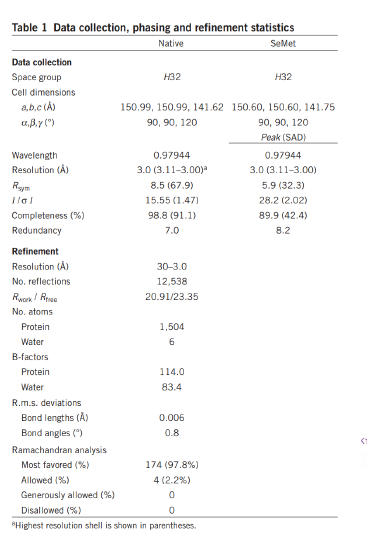
위 테이블의 의미를 알아야하지요
원자의 구조와 본딩 사이의 표준적인 거리로 화학 구조를 대충 유추할 수 있다.
단백질은 원자의 위치와 대칭성, 구조가 어느 정도 정해져 있다.
space group이 뭐지.
단백질에서 어떤 신호가 대칭적으로, 반복적으로 나오는지 확인한다.
cell dimensions가 뭐지. 격자 크기인가
wavelangth, x선의 wavelength. 격자만한 길이여야 한다.
resolution은 해상도. 구분할 수 있는 거리.
x선 결정학의 발견, 화절에 의한 결절 무늬가 일정하게 나온다. Max von Laue가.
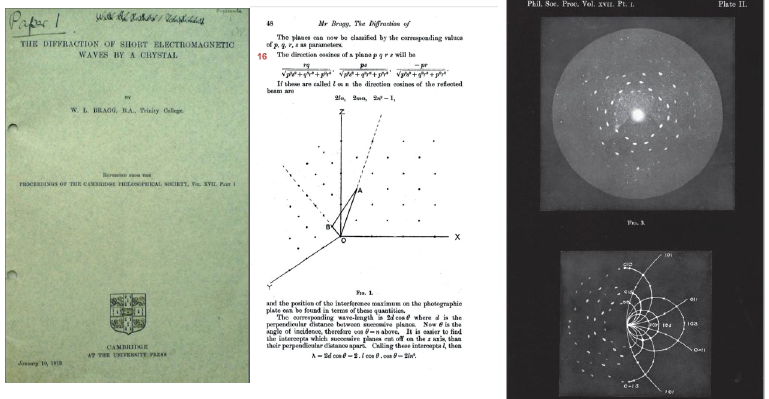
Bragg 부자가 회절 무늬를 격자 구조로 해석하는 방법을 알아냈다.
X선은 원자와 충돌한 후 탄성 충돌해야 해. 빛은 파장이어야 해.
삼각함수를 알아야 하지요호
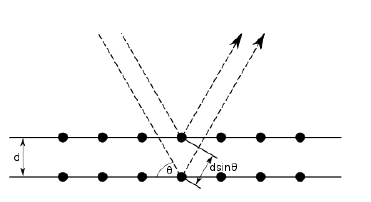
Abbel 공식
분자의 원자 위치를 xyz 3차원 공간의 좌표로 찍어.
원자의 종류와 좌표값, 네 가지 정보.
원자 사이 연결성을 나타내기 위한 번호 라벨링도 필요.
이런 분자 구조의 모든 정보를 protain data라고 부른다.
Atomic resolution structure의 의미가 무엇인가?

원자 사이 화학 반응을 알 수 있을 정도로 원자 사이 거리를 알 수 있어야 한다.
원자단위로 돌연변이를 만들고, 그 결과를 예측할 수 있을 정도로 알아야 한다.
왜 이런 걸 알아야 하는가?
- mechanism 제안
- 반응 모델 제안
- 신약 설계
- structural genomics
단점도 많은 X-ray 결정학

일단 결정을 만들어야 한다.
두 가지 종류의 상태가 바뀌는 현상을 볼 수 없다.
그나마 좋은 점은 크기 제한이 없다는 것. 그러나 너무 크면 결정을 만들기 힘들긴 하다.
철 설레늄 같은 독특한 원자의 위치를 알기 쉽다.
그러나 수소는 특정할 수 없다. x선이 뚫고 지나가기 때문이다.
Cryo EM은 큰 걸 보기 좋다.
EM은 모든 원자를 볼 수 있다.
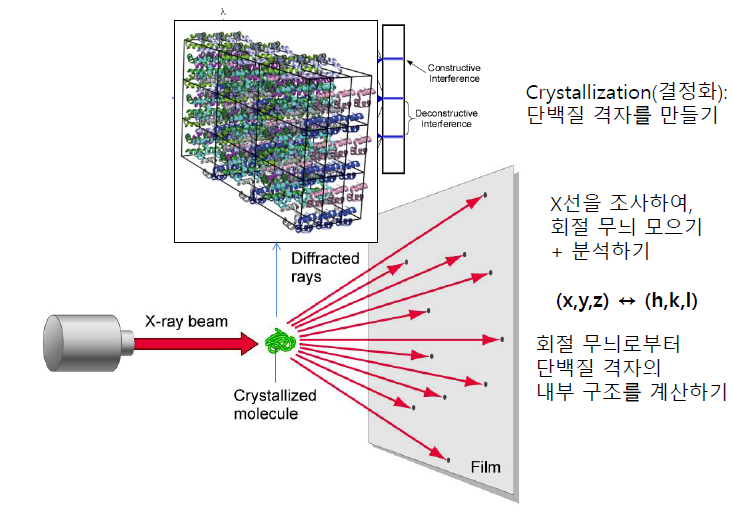
상자(lattice) 하나당 단백질 하나.
k-space와 rea-space의 매치
X 레이 광원이 발달해
4세대까지 발달.
x선을 계속 쬐이지 않고 펄스를 줘

3차원으로 돌려서 입체 정보를 얻어.
푸리에에에에에에엥 변환
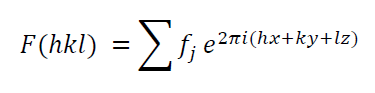
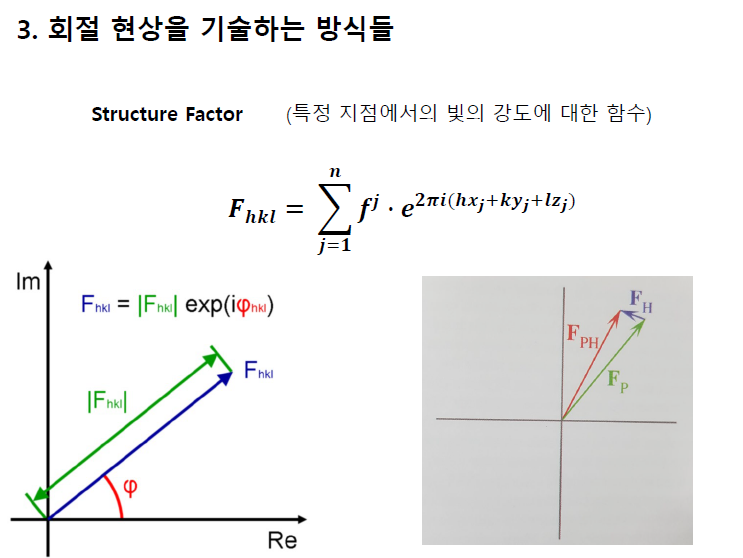
structure factor를 구하는 방법

이 공식을 꼭 알아라.
structure factor가 뭐더라.
이게 뭔지 꼭 알아야 한다.
ccd 센서는 phase를 인식하지 못 한다. 어쩌지? 따로 감지한다.

위 키워드가 모두 시험 문제다.
Ewald sphere을 그릴 수 있으면 좋다. 이게 쉽게 설명하기 위해 있는 모델이래.
R-factor가 중요하대. 잘 알고 있어래. 논문의 질을 판단할 수 있기 때문에.
CC 결합을 기점으로 하면 1.54옹스트롬.
2옹스트롬 정도면 좋은 해상도이다.
왜 단백질 결정을 만들어야 하는가?
어떻게 대칭적이지 않은 단백질로 결정을 만드는 가?
unit cell안에서는 대칭적으로 있기 때문.
가능한 unit cell의 수가 많아야 회절이 잘 되고 간섭이 잘 되고 무늬가 강해져
격자가 틀어지면 데이터가 잘 안 뽑힘
관측값으로 유추한 단백질 구조와 실제 구조를 어떻게 비교하나?
어떻게 fitting이 잘 되었는지 확인하나?
유추한 단백질이 만드는 diffraction pattern과 실제 패턴을 비교하고 R value를 구한다.

또또 이 수식이 나온다.
알아야 한다. 외워야 한다. 문제다!
F 는 structure factor, 패턴의 관측값
rho는 밀도 함수.
양측은 푸리에 변환 관계
왜 식을 알아야 하는가?

대가가 왜 5개의 논문을 철회했는가?
코드에 - 부호를 잘못 넣어서 단백질의 상이 거울상으로 나왔다는 웃픈 이야기.
또또 회절 기초 이야기
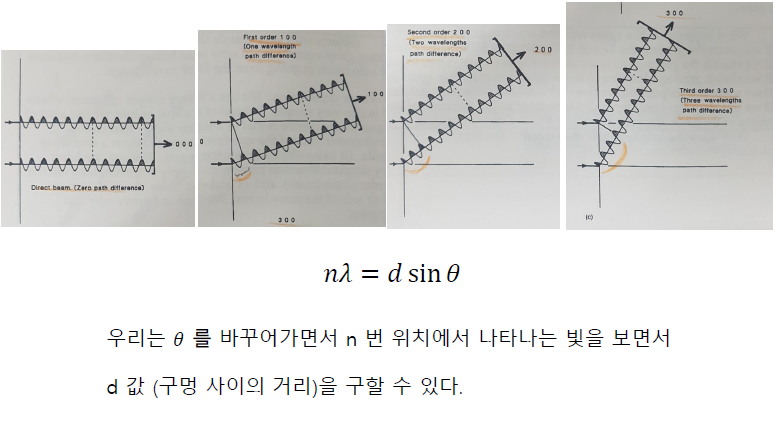
h m l 격자수를 사용

아니 이게 왜 나오는 식이지
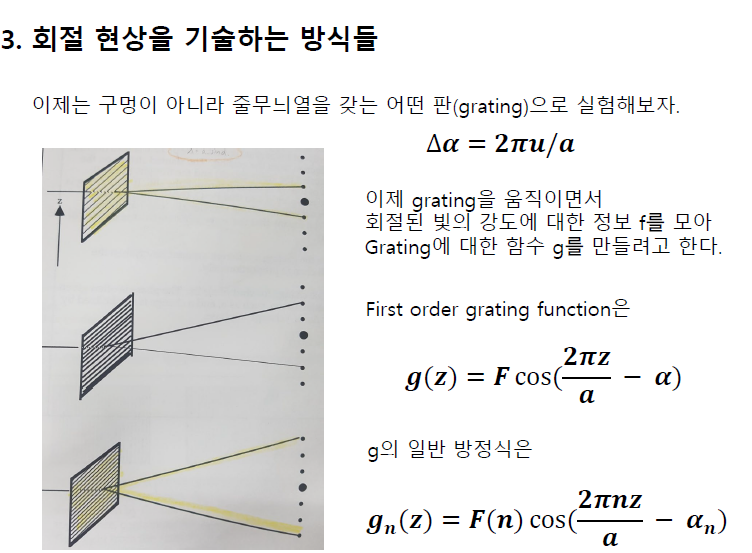
grating function의 의미가 뭔가요?
패턴을 이용해서 역으로 격자의 모양을 구해보겠다.

그러니까 이 식을 disctized Fourier transform으로 봐야 한다.
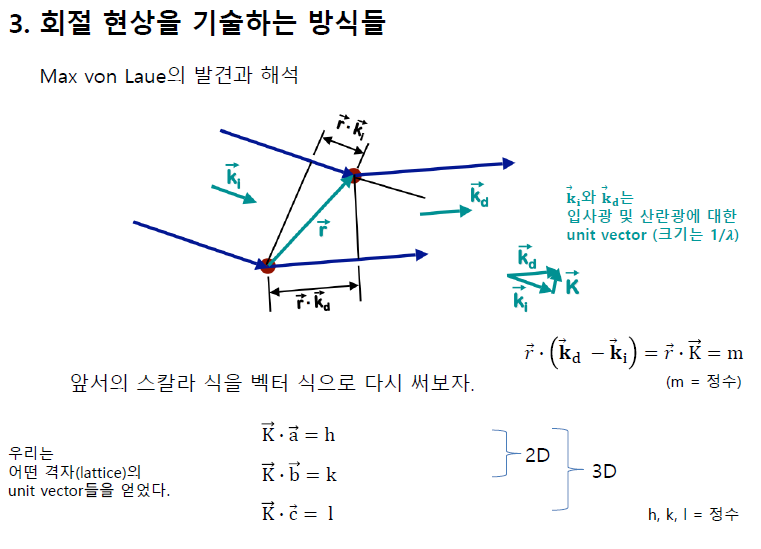
hkl이 뭘 뜻하는 거였나? 몇 번째 격자였나?

저 스타 붙은 게 격자 면에 대한 벡터
스타는 reciprocal space에 대한 unit vector
실수계에서 격자 사이의 간격이 좁아지면 회절 무늬에서 간격을 늘어난다.
아이고 고체 물리 다시 공부해야겠네.
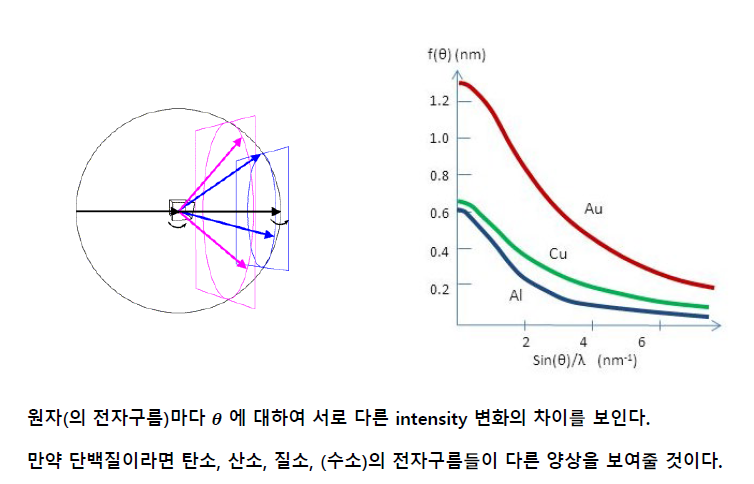
원자마다 빛의 산란 각도마다 나오는 빛의 intensity가 정해져있다. 이걸

왜 정수배의 파수만 나오는가? → 보강간섭이기 떄문
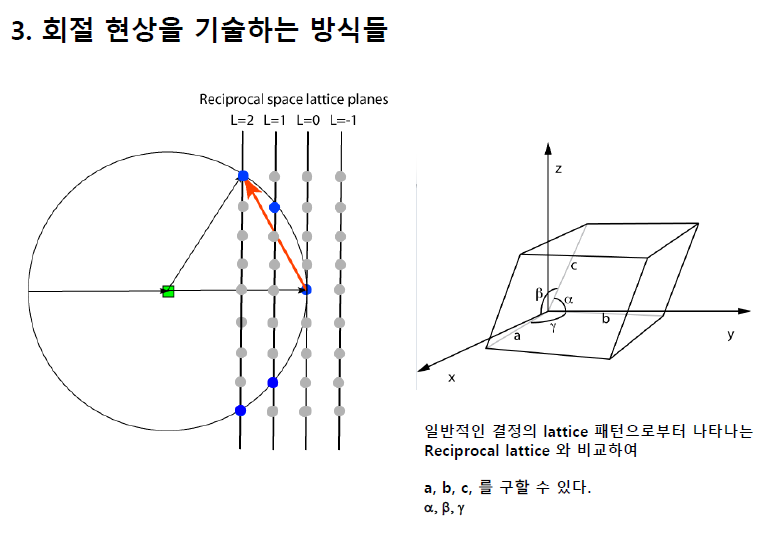
스크린이 가까워질 수록, 더 큰 파수의 값이 나오고, 이것은 real space에서는 더 작은 공간의 정보를 담는다.
2차원 푸리에 변환에서 변두리에 있는(큰 파수의) 정보는 real space에서 작은 디테일에 관한 정보일 것이다.
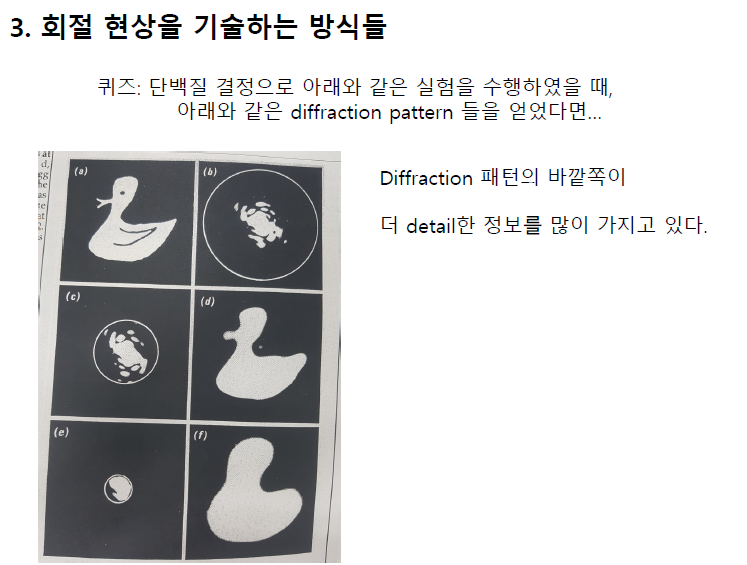

위상에 대한 정보가 날아간 상태.
역변환을 위해서는 위상에 대한 정보가 필요하다.
intensity는 격자의 모양과 원자로 결정.
원자를 바꾸면
원자를 살짝 바꾸면
이러면 역으로 위상에 대한 정보를 알 수 있다.
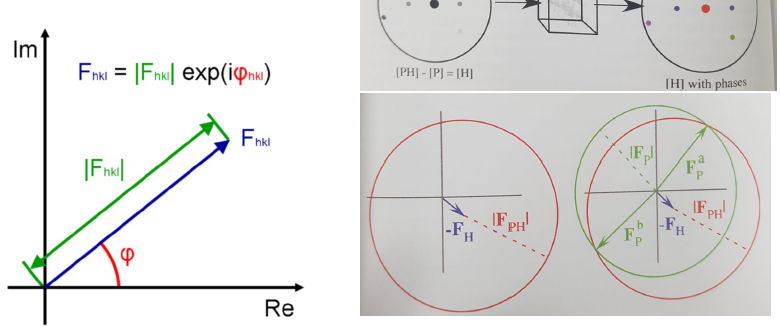
그런데 heavy atom을 어떻게 넣죠?
수은같은 원자가 잘 붙는 residue가 따로 있다고 한다.
넣어도 단백질 모양이 변하지 않는다고 한다.
heavy atom을 여러개 쓰면 더 위상을 특정할 수 있다.
더 똑똑하게 heavy atom을 붙이는 방법
seleno-methionine labeling을 사용한다.

특수한 minimum 배지를 사용. 아미노산 합성을 새로 못 하게. 밖에서 만들어준 아미노산만 사용하게.
다른 단백질에서 얻은 패턴을 이용해서 phase를 알아낼 수 있지 않을까?패턴 사이의 차이점을 이용해서 구조의 차이점을 알아내자. 이 차이는 위상에 의해 만들어진 거라고 여기고 계산하자.
단백질의 패턴을 비교하려면 이리저리 상을 회전하고 변환해야 한다. 회전과 위상에 의한 matrix를 구하는 것이다.
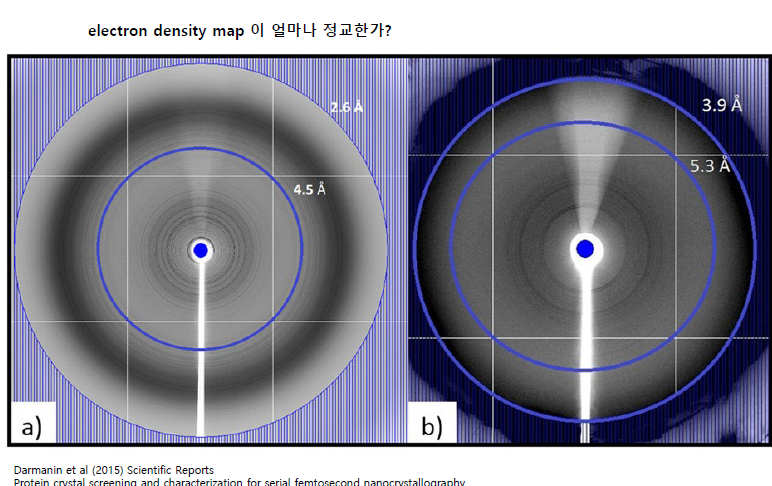
최대한 바깥쪽의 정보를 긁어 와서 해상도를 높이자.

1번은 유추한 구조가 만드는 패턴과 실제 패턴의 오차를 구하는 것.
이것보다는 더 부드러운 오류 측정법

R value를 이용한다.
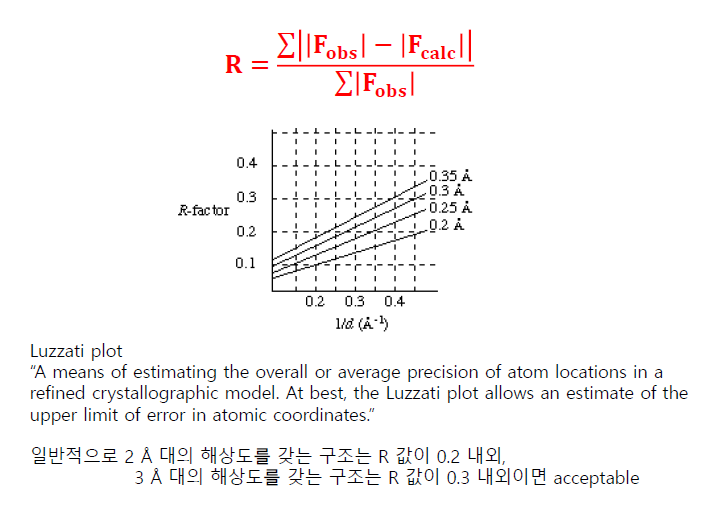
해상도에 의해 R 값이 제한.

제일 힘든 부분, 단백질 결정 만들기, 진짜 어렵다
단백질은 흐물흐물,
차곡차곡 쌓이는 게 힘들다. 그래서 결정이 좀 삐뚤삐뚤 → 보강간섭 약화
결정 자체는 잘 만들었는데, cell안에서 단백질의 모양이 다양하게 다를 수 있다.
결정을 어떻게 만드나?
아주 진한 농도로 단백질 물을 만든다. 그게 스스로 침전된다.
salting out으로 인한 hydrophobic interaction
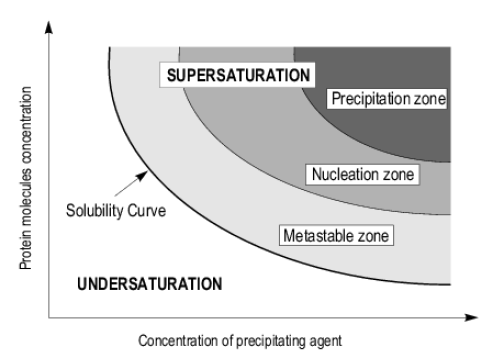
precipitating agent(결정화를 도와주는 애들?)
단백질 결정은 precipiation zone 수준이 아니다.
핵을 만들기 위해서는 nucleation zone과 precipitation zone 에 걸터있는 수준이다.
결정 핵 주위로 단백질이 뭉치려면 metastable zone에 있어야 한다.
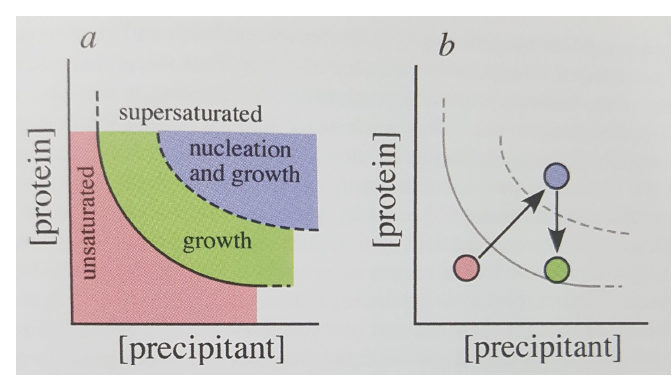
파랑에서 초록으로 가는 ㅇ경로가 아랫방향인 이유는 단잭질을 먹어 치우면서 농도가 낮아졌기 때문.
단백질 농도를 높이는 가장 쉬운 방법은 증발. 그것도 아주 서서히.
hangging drop방식과 sitting drop 방식.
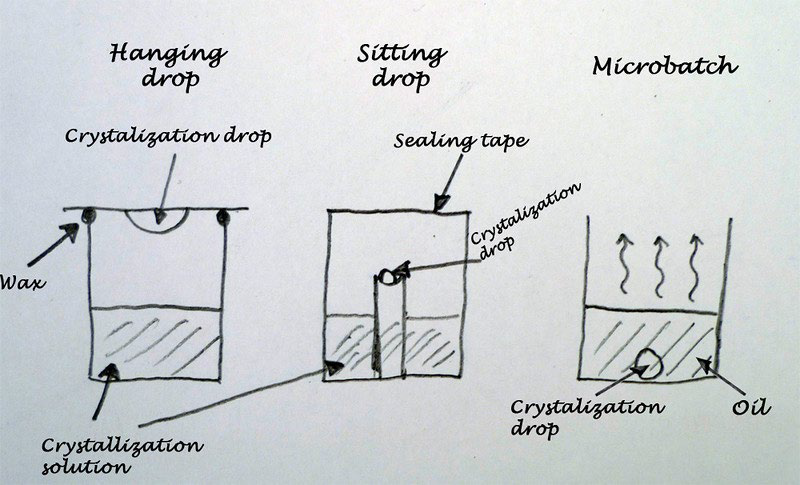
오만가지를 변화해서 만들어 볼 수 있어요
하나라도 맞아라.
어떻게 하면 팔랑거리는 부위를 딱딱하게 만들까.
항체(nanobody)를 달아 버릴까?
결정을 잘 만드는 lysozyme을 달아버릴까?
Ligand 넣어버리면 유연성이 좀 줄어들지 않을까

이거 시험 문제다.
x선 결정학은 이제 가는 해다.
Cryo-EM은 이제 상승세다.